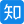ViSNet:用于分子性质预测和动力学模拟的通用分子结构建模网络
2024-01-31 | 作者:科学智能中心
编者按:尽管几何深度学习已经彻底颠覆了分子建模领域,但最先进的算法在实际应用中仍然面临着几何信息利用不足和高昂计算成本的阻碍。为此,微软研究院科学智能中心(Microsoft Research AI4Science)的研究员们提出了通用分子结构建模网络 ViSNet。在多个分子动力学基准测试中,ViSNet 均表现优异。
分子几何建模在理解生物活性机制、化学性质预测、药物设计和蛋白质工程方面发挥着关键作用。然而,虽然几何深度学习(geometric deep learning)是一种低成本、高精度且可以被广泛使用的计算方法,在过去十年取得了巨大进展,但这种技术仍然存在一些有待解决的问题和局限性:
- 分子可解释性不足:深层神经网络尽管可以进行预测,但缺乏对分子的深入洞察;
- 随着分子尺寸的增加,计算成本迅速增加:一些目前最先进的方法中采用的高阶 Clebsch-Gordan 系数计算是计算密集型的,因此阻碍了其在大分子中的应用;
- 需要实际应用中的盲目测试和评估:模型总是在基准测试上进行测试,同时也需要仔细评估在实际应用中的有效性。
为了解决这些难题,微软研究院科学智能中心的研究员们将研究重点聚焦在了如何提高分子可解释性、降低计算成本以及评估实际应用几个方面,并创新性地提出了通用分子结构建模网络 ViSNet (Vector-Scalar interactive graph neural Network)。相关文章“Enhancing geometric representations for molecules with equivariant vector-scalar interactive message passing”已发表在《自然-通讯》(Nature Communications)杂志上,并同时入选了“AI and machine learning”和“Biotechnology and method”两个领域的编辑精选文章。
ViSNet论文链接:https://www.nature.com/articles/s41467-023-43720-2
“AI and machine learning”领域编辑精选文章链接:https://www.nature.com/collections/ceiajcdbeb
“Biotechnology and method”领域编辑精选文章链接:https://www.nature.com/collections/idhhgedgig
有效提升分子几何表示
研究员们最初计划通过有效且充分地利用分子结构的领域知识来设计模型。由于经典分子动力学(molecular dynamics, MD)通过明确描述势能函数中的键长、键角和二面角来模拟分子运动,所以受经典 MD 模拟的启发,研究员们将这些项目转换并扩展,从而构建了 ViSNet 独特的模型设计。
与通过简单的特征工程过程直接采用角度或二面体信息不同,研究员们提出了“方向单元”这个概念作为节点的向量化表示,即从中心节点到其任何第一个相邻节点的所有归一化向量的总和,作为中心节点的矢量化表示。再以此将键长、键角和二面角计算扩展到二体、三体和四体相互作用。然后,通过设计运行时几何计算(runtime geometry calculation, RGC)模块来描述模型操作等多体交互。
更重要的是,三体和四体相互作用的 RGC 计算都只有线性时间复杂度。因此,研究员们又进一步提出了向量标量交互式消息传递机制(ViS-MP),其中方向单元会通过构建块由节点和边的标量表示迭代更新,反过来,标量表示由方向单元同时更新 RGC 模块。RGC 和 ViS-MP 的独特设计显著增强了几何编码能力并加速了分子图神经网络中的消息传递过程。
图1:ViSNet 网络结构示意图
ViSNet在分子建模和性质预测实际应用中的表现
研究员们首先将 ViSNet 在广泛使用的分子化学性质预测基准上进行了评估。在 MD17、修订版 MD17 、 MD22、QM9 以及 Molecule3D 数据集上显示出卓越的性能,证明了分子几何表示的强大能力。然后,研究员们还在自已开发的 DFT(密度函数理论)精度的蛋白质数据集 AIMD-Chig 数据集上训练了 ViSNet,并对蛋白质 Chignolin 进行了 MD 模拟。
ViSNet 取得了比经验力场和现代机器学习力场更好的性能及令人满意的结果。ViSNet 的模拟结果与在 DFT 水平上获得的结果非常接近,这表明 ViSNet 在数据效率和模拟保真度方面具有潜力。
研究员们用 ViSNet 参加了全球首届 AI 药物研发算法大赛。该大赛旨在根据小分子的序列信息(即SMILES)预测针对新冠病毒 SARS-CoV-2 主要蛋白酶的抑制剂。共有来自全球878支团队的1105名参赛者参与了此次比赛。最终,研究员们凭借 ViSNet 获得了比赛的总冠军,也展现了 ViSNet 优异的预测准确性。
如何获取ViSNet模型?
为了促进更广泛的应用和便捷的使用,ViSNet 已被微软纳入 Pytorch Geometry 库,作为分子建模和属性预测领域的基本模型。ViSNet 的定期维护和更新版本也可在 GitHub上 获取。
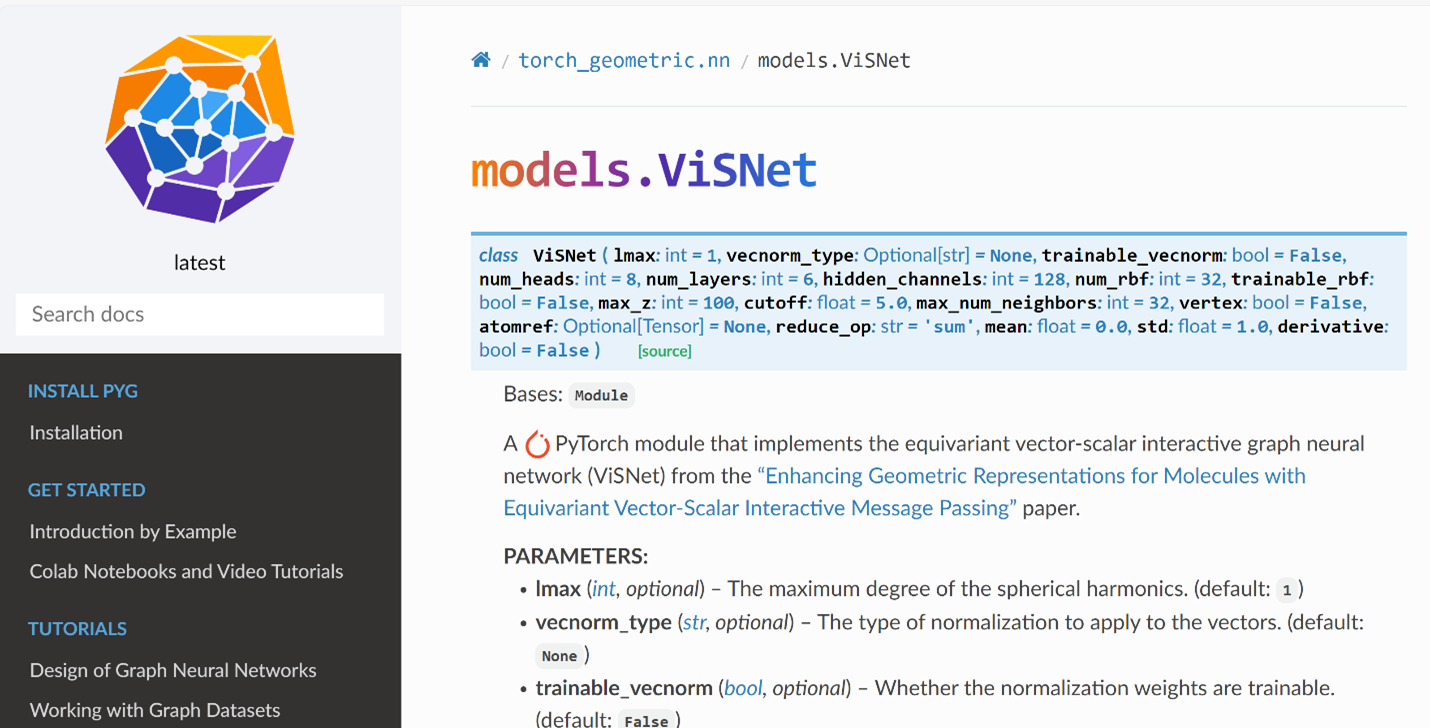
图2:ViSNet 在 Pytorch Geometry 中作为基础模型
Pytorch Geometry 库链接:https://pytorch-geometric.readthedocs.io/en/latest/generated/torch_geometric.nn.models.ViSNet.html
GitHub 链接:https://github.com/microsoft/AI2BMD/tree/ViSNet
此外,考虑到图神经网络随着模型变得越来越大、越来越深,可能会遇到“过度平滑”的风险,研究员们还进一步设计了 ViSNet 的 Transformer 版本,可以将 RGC 模块转移到 Transformer 注意力计算中,并提出了一种新颖的原子间位置编码(IPE),命名为 Geoformer(Geometric Transformer的缩写)。作为 ViSNet 的 Transformer 变体,Geoformer 可通过堆叠数百个注意力块来进行大模型训练。相关研究论文发表于 NeuraIPS 2023。
分子动力学模拟的未来:兼具人工智能与从头计算精度的能力
作为人工智能(AI)驱动的从头算分子动力学(AI2BMD)项目的重要组成部分,ViSNet 致力于实现加速分子动力学模拟的目标,使大型分子系统的模拟精度接近从头算法。
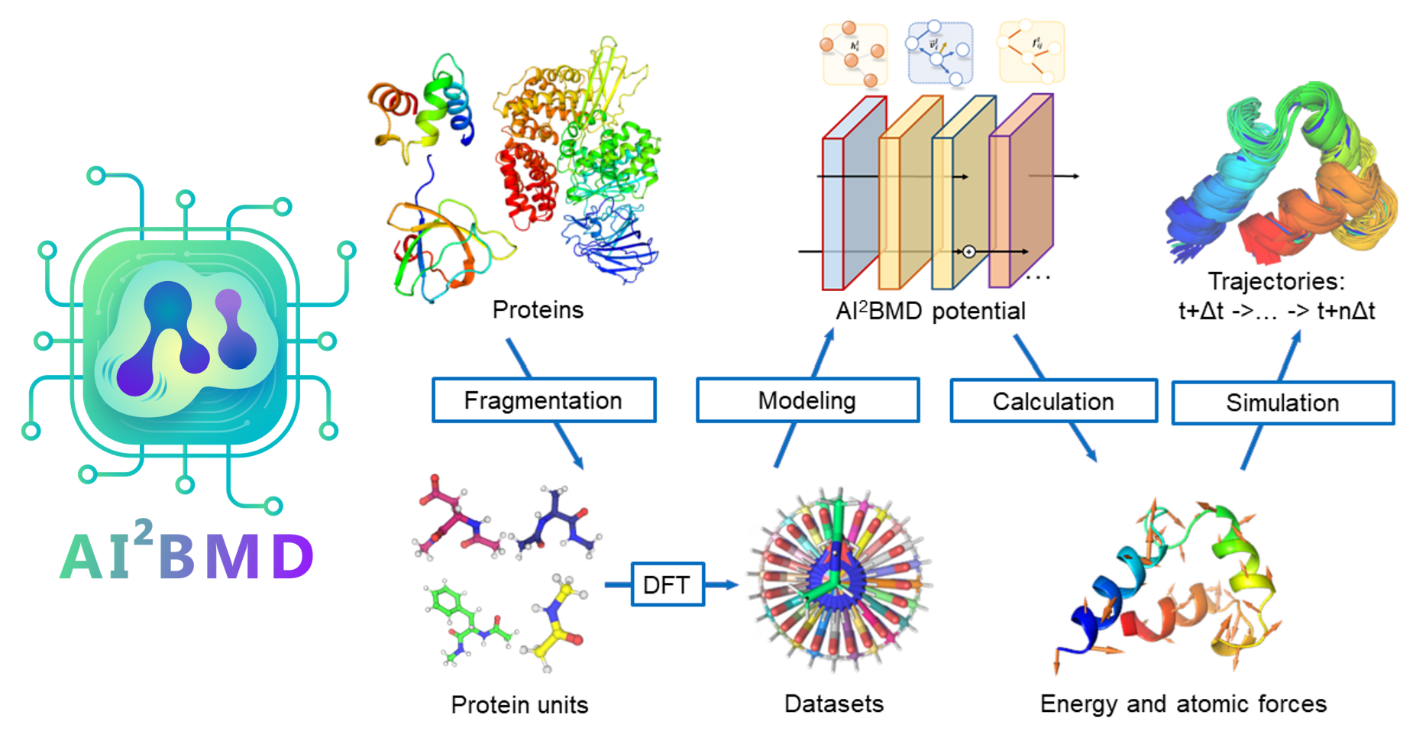
图3:AI2BMD 整体流程
ViSNet 可以让 AI2BMD 实现对包含超过10,000个原子的蛋白质的能量和力计算达到接近从头算法的精度。利用 ViSNet 进行蛋白质动力学模拟还可提高自由能估计的准确性,提供有关蛋白质折叠热力学的深入预测,并有助于表征蛋白质的特性,从而潜在地增强实验研究。
相关链接:
ViSNet论文:https://www.nature.com/articles/s41467-023-43720-2
AIMD-Chig 数据集:https://www.nature.com/articles/s41597-023-02465-9
首届AI药物研发算法大赛官方网页:https://aistudio.baidu.com/competition/detail/1012/0/leaderboard
ViSNet-Pytorch Geometry 库:https://pytorch-geometric.readthedocs.io/en/latest/generated/torch_geometric.nn.models.ViSNet.html
ViSNet-GitHub:https://github.com/microsoft/AI2BMD/tree/ViSNet
Geoformer:https://github.com/microsoft/AI2BMD/blob/Geoformer/Geoformer.pdf/
AI2BMD:https://microsoft.github.io/AI2BMD/index.html